Pipeline parameters¶
PhyloNext includes numerous configurable parameters.
For convenience, they are grouped into several categories:
- Input/output options
- Data subsetting:
- Taxonomic scope
- Spatial scope
- Spatial outliers removal
- Occurrence filtering and binning
- Diversity estimation
- Visualization
- Interactive (Leaflet-based)
- Static (maps in pdf format)
- Phylogenetic tree-related parameters
- Generic options
- Nextflow-specific parameters
Input/output options¶
Define where the pipeline should find input data and save output data.
| Parameter | Description | Type | Default |
|---|---|---|---|
--input |
Path to the directory with parquet files (Parquet format) 1 | directory |
|
--outdir |
The output directory where the results will be saved | directory |
./results |
--phytree |
Custom phylogenetic tree in Newick format (optional) 2 | file |
1: GBIF occurrence dump in the Parquet format. Could be stored locally or in the cloud (S3 or Azure Blob storage).
2:
Users have the option to provide their own custom phylogenetic tree.
If this option is chosen, the tips of the phylogenetic tree should be labeled
either with Latin binomials (for example, "Homo_sapiens"),
or with Open Tree IDs (for example, "ott359899").
Be sure to adjust the --phylabels parameter accordingly.
Additionally, a set of phylogenetic trees, which come with descriptions and
are pre-packaged with the pipeline, are available at this link:
https://github.com/vmikk/PhyloNext/tree/main/test_data/phy_trees
Taxonomic scope¶
Define which taxa should be analyzed.
| Parameter | Description | Type | Example | Default |
|---|---|---|---|---|
--phylum |
Phylum to analyze 1 | string |
"Chordata" | |
--classis |
Class to analyze 1 2 | string |
"Mammalia" | |
--order |
Order to analyze 1 | string |
"Carnivora" | |
--family |
Family to analyze 1 | string |
"Felidae,Canidae" | |
--genus |
Genus to analyze 1 | string |
"Felis,Canis,Lynx" | |
--specieskeys |
Custom list of GBIF specieskeys (text file with a single column) 3 | file |
"SpeciesKeys.txt" | |
--noextinct |
File with extinct species specieskeys for their removal (file with a single column, with header) | file |
"Carnivora" | |
--excludehuman |
Exclude genus "Homo" from occurrence data | boolean |
True | True |
1: Multiple comma-separated values allowed (e.g., "Felidae,Canidae" for the Family rank).
2:
Unfortunately, class is a reserved keyword in Nextflow.
Therefore, Latin classis is used as a parameter name.
3:
The --specieskeys argument enables users to specify a custom list of GBIF species keys.
This should be provided as a text file containing a single column of species keys,
each corresponding to a specific species as defined in the GBIF taxonomic backbone
(https://www.gbif.org/species/search).
These species keys can belong to various taxa, including non-monophyletic clades,
and are used to filter and retrieve specific species data from GBIF.
Example of the file:
Spatial scope¶
Spatial filters.
| Parameter | Description | Type | Example | Default |
|---|---|---|---|---|
--latmin |
Minimum latitude of species occurrences 1 | number |
5.1 | |
--latmax |
Maximum latitude of species occurrences 1 | number |
15.5 | |
--lonmin |
Minimum longitude of species occurrences 1 | number |
47.0 | |
--lonmax |
Maximum longitude of species occurrences 1 | number |
55.5 | |
--country |
Country code, ISO 3166 format 2 | string |
"DE,PL,CZ" | |
--polygon |
Custom area of interest (a file with polygons in GeoPackage format) | file |
Brazil_ThreeStates.gpkg |
|
--wgsrpd |
Polygons of World Geographical Regions 3 | file |
pipeline_data/WGSRPD.RData |
|
--regions |
Names of World Geographical Regions 4 | string |
"L1_EUROPE,L1_ASIA_TEMPERATE" |
1: Coordinates should be provided in decimal degrees
2:
Country codes should be provided in the two-letter ISO 3166-1 alpha-2 coding system,
see details here (column Alpha-2 code),
and this Wikipedia article.
3:
PhyloNext ships WGSRPD shapefile as a built-in data.
To use it, specify a full path to the data (e.g., $(realpath "${HOME}/.nextflow/assets/vmikk/PhyloNext/pipeline_data/WGSRPD.RData"))
4: Multiple comma-separated values allowed.
Currently, Level-1 and Level-2 WGSRPD polygons are supported.
Level-1 polygons (continental level):
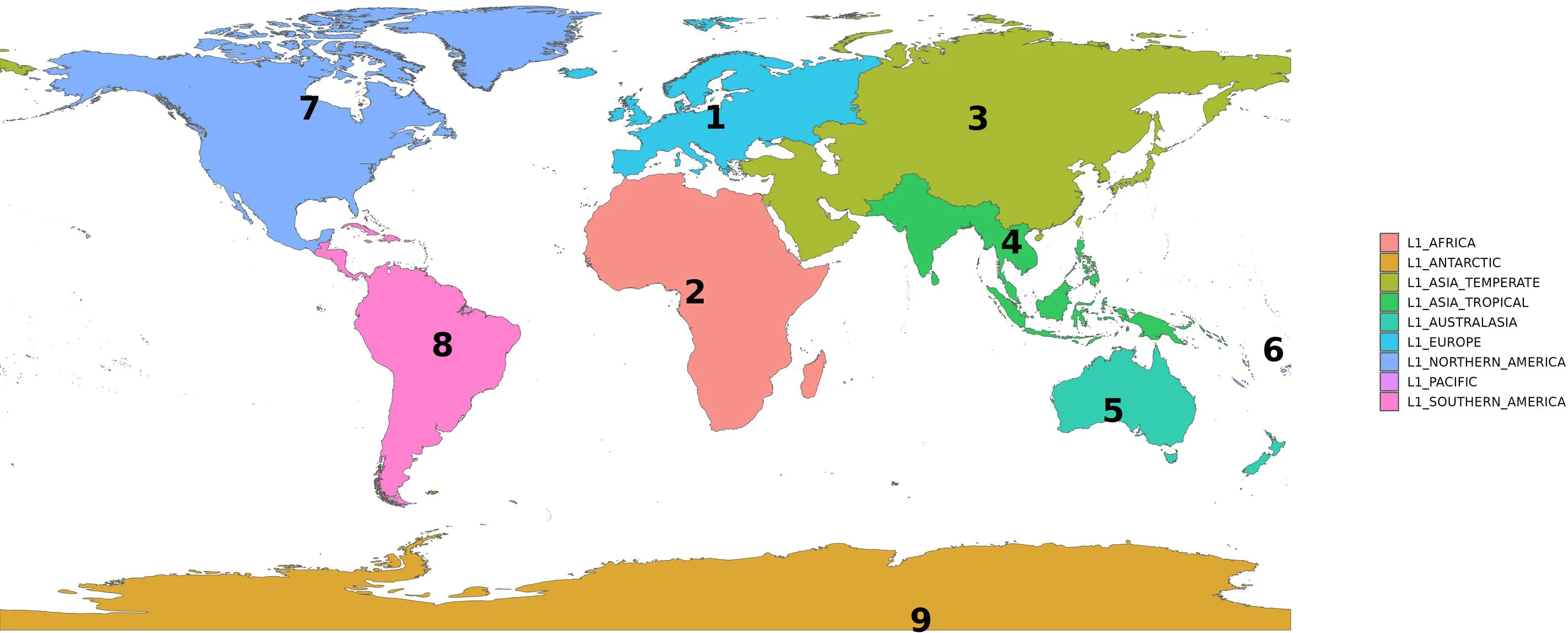
Level-2 polygons (regional or subcontinental level):
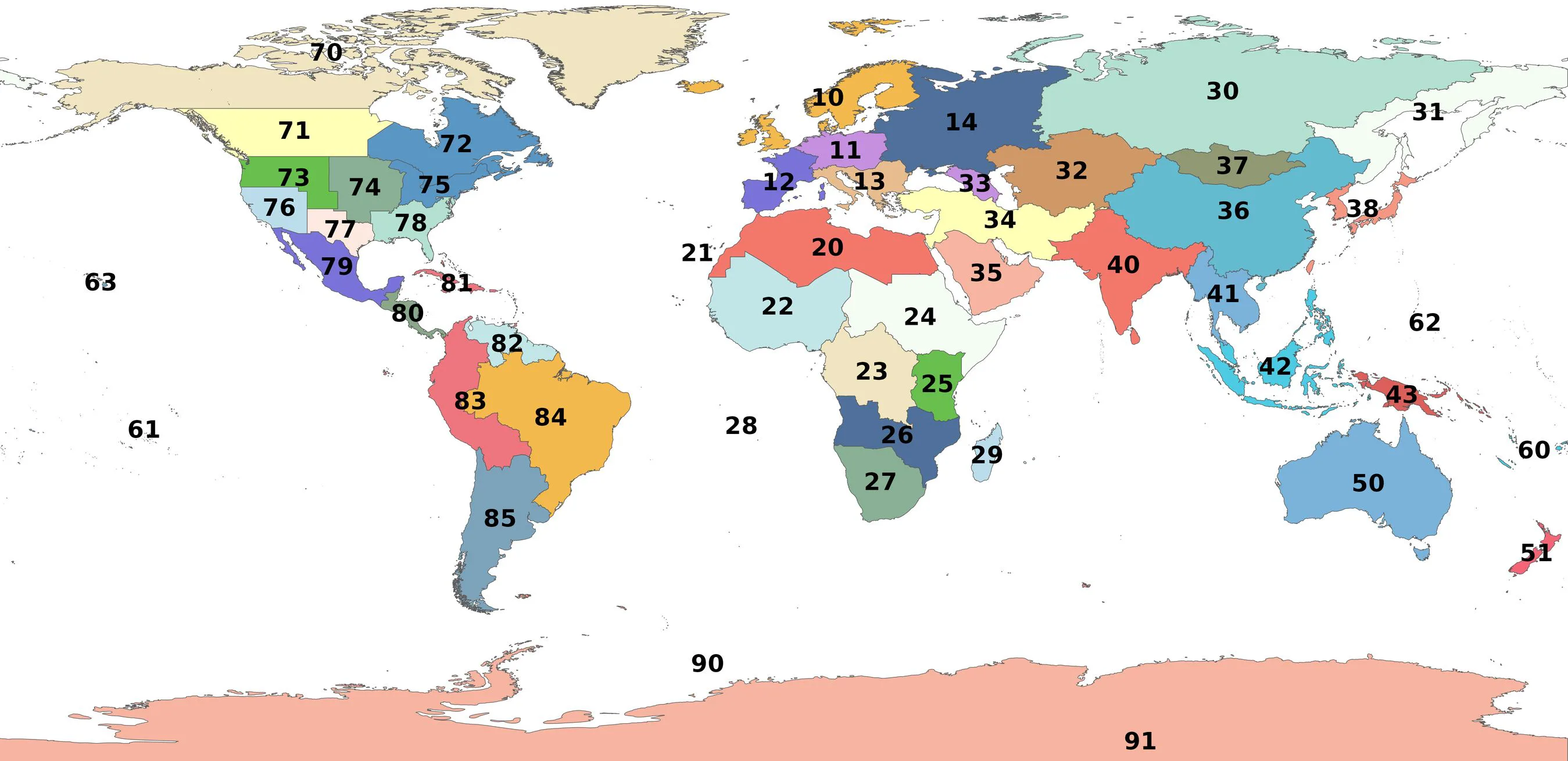
| Number | Polygon code |
|---|---|
| 1 | L1_EUROPE |
| 2 | L1_AFRICA |
| 3 | L1_ASIA_TEMPERATE |
| 4 | L1_ASIA_TROPICAL |
| 5 | L1_AUSTRALASIA |
| 6 | L1_PACIFIC |
| 7 | L1_NORTHERN_AMERICA |
| 8 | L1_SOUTHERN_AMERICA |
| 9 | L1_ANTARCTIC |
| ------ | --------------------------------- |
| 10 | L2_Northern_Europe |
| 11 | L2_Middle_Europe |
| 12 | L2_Southwestern_Europe |
| 13 | L2_Southeastern_Europe |
| 14 | L2_Eastern_Europe |
| 20 | L2_Northern_Africa |
| 21 | L2_Macaronesia |
| 22 | L2_West_Tropical_Africa |
| 23 | L2_West_Central_Tropical_Africa |
| 24 | L2_Northeast_Tropical_Africa |
| 25 | L2_East_Tropical_Africa |
| 26 | L2_South_Tropical_Africa |
| 27 | L2_Southern_Africa |
| 28 | L2_Middle_Atlantic_Ocean |
| 29 | L2_Western_Indian_Ocean |
| 30 | L2_Siberia |
| 31 | L2_Russian_Far_East |
| 32 | L2_Middle_Asia |
| 33 | L2_Caucasus |
| 34 | L2_Western_Asia |
| 35 | L2_Arabian_Peninsula |
| 36 | L2_China |
| 37 | L2_Mongolia |
| 38 | L2_Eastern_Asia |
| 40 | L2_Indian_Subcontinent |
| 41 | L2_Indo_China |
| 42 | L2_Malesia |
| 43 | L2_Papuasia |
| 50 | L2_Australia |
| 51 | L2_New_Zealand |
| 60 | L2_Southwestern_Pacific |
| 61 | L2_South_Central_Pacific |
| 62 | L2_Northwestern_Pacific |
| 63 | L2_North_Central_Pacific |
| 70 | L2_Subarctic_America |
| 71 | L2_Western_Canada |
| 72 | L2_Eastern_Canada |
| 73 | L2_Northwestern_USA |
| 74 | L2_North_Central_USA |
| 75 | L2_Northeastern_USA |
| 76 | L2_Southwestern_USA |
| 77 | L2_South_Central_USA |
| 78 | L2_Southeastern_USA |
| 79 | L2_Mexico |
| 80 | L2_Central_America |
| 81 | L2_Caribbean |
| 82 | L2_Northern_South_America |
| 83 | L2_Western_South_America |
| 84 | L2_Brazil |
| 85 | L2_Southern_South_America |
| 90 | L2_Subantarctic_Islands |
| 91 | L2_Antarctic_Continent |
Custom polygons
It is possible to use custom polygons instead of WGSRPD file.
Polygons should be in the simple feature collection format (class sf of the sf package,
geometry type: MULTIPOLYGON) and must contain LevelName column.
Data should be saved as a serialized R object (with saveRDS).
Spatial outliers removal¶
| Parameter | Description | Type | Example | Default |
|---|---|---|---|---|
--dbscan |
Remove spatial outliers with density-based clustering | boolean |
True | False |
--dbscannoccurrences |
Minimum species occurrence to perform DBSCAN | integer |
30 | 30 |
--dbscanepsilon |
DBSCAN parameter epsilon, km | integer |
1500 | 1500 |
--dbscanminpts |
DBSCAN min number of points | integer |
3 | 3 |
Currently, only density-based clustering (DBSCAN) algorithm is implemented for removal of spatial outliers.
The DBSCAN algorithm requires 2 parameters:
- eps (--dbscanepsilon), which specifies how close points should be to each other to be considered a part of a cluster
If the distance between two points is lower or equal to this value, these points are considered neighbors.
- minPoints (--dbscanminpts), the minimum number of points to form a dense region.
Parameter --dbscannoccurrences is used to skip DBSCAN filtering for species with low number of unique points (e.g., <30).
For more details, see the blog post "Outlier Detection Using DBSCAN" by John Waller.
Occurrence filtering and binning¶
| Parameter | Description | Type | Example | Default |
|---|---|---|---|---|
--minyear |
Minimum year of record's occurrences | integer |
2000 | 1945 |
--maxyear |
Maximum year of record's occurrences | integer |
2010 | |
--coordprecision |
Coordinate precision threshold, decimal degrees 1 | number |
0.1 | 0.1 |
--coorduncertainty |
Maximum allowed coordinate uncertainty, meters 1 | number |
10000 | 10000 |
--coorduncertaintyexclude |
Black list of coordinate uncertainty values 1 | string |
"9999" | "301,3036,999,9999" |
--basisofrecordinclude |
Round spatial coordinates to N decimal places 2 | string |
"PRESERVED_SPECIMEN" | |
--basisofrecordexclude |
Round spatial coordinates to N decimal places 2 | string |
"FOSSIL_SPECIMEN" | "FOSSIL_SPECIMEN,LIVING_SPECIMEN" |
--h3resolution |
Spatial resolution of the H3 geospatial indexing system | integer |
4 | 4 |
--roundcoords |
Round spatial coordinates to N decimal places 3 | integer |
2 | 2 |
For spatial binning of species occurrences, PhyloNext uses H3 geospatial indexing system developed by Uber.
H3 represents a hierarchical geospatial index, where each hexagonal grid cell has a unique index (e.g., 8a1f05835a37fff for GBIF headquarter).
H3 supports 16 resolutions (from 1 to 16), which can be selected in PhyloNext using --h3resolution parameter.
By default, PhyloNext uses resolution 4, which corresponds to a hexagon with edge length of 22.6 km and cell area of 1170 km2.
More details on H3 resolutions could be found here.
1:
It's possible to remove occurrence records with a high level of coordinate uncertainty or low precision.
It can be done using some threshold values.
Also, there are several known default values for coordinate uncertainty in meters, which could be black-listed as well
(e.g., these values may correspond to records linked to country centroids).
NB! Records with missing values will not be removed, as many publishers do not fill these fields in the database.
For details, see the blog post. "Common things to look out for when post-processing GBIF downloads" by John Waller.
2:
For details, see description of a Darwin Core term Basis of record
and a short explanation here.
Multiple comma-separated values allowed.
3:
As DBSCAN filtering is very computationally intensive,
it is possible to reduce dataset size (almost without loosing precision) by rounding record coordinates.
By default, coordinates are rounded to 2 decimal places,
which corresponds to an accuracy of ~1.11 km at the equator.
To disable coordinate rounding, set --roundcoords to a negative values (e.g., -1).
Removal of common spatial errors¶
PhyloNext implements several filters analogous to the filters
in CoordinateCleaner R package by Alexander Zizka:
- Country and province centroids
- Capital coordinates
- Coordinates of biodiversity institutions
- Urban areas
- Seas
| Parameter | Description | Type | Default |
|---|---|---|---|
--terrestrial |
Land polygon for removal of non-terrestrial occurrences | file |
enabled |
--rmcountrycentroids |
Polygons with country and province centroids | file |
disabled |
--rmcountrycapitals |
Polygons with country capitals | file |
disabled |
--rminstitutions |
Polygons with biological institutions and museums | file |
disabled |
--rmurban |
Polygons with urban areas | file |
disabled |
PhyloNext provides files for removal of common spatial errors,
the data are built-in in the pipeline_data direcory and can be selected in the following way:
--terrestrial `$(realpath "${HOME}/.nextflow/assets/vmikk/PhyloNext/pipeline_data/Land_Buffered_025_dgr.RData")`
--rmcountrycentroids `$(realpath "${HOME}/.nextflow/assets/vmikk/PhyloNext/pipeline_data/CC_CountryCentroids_buf_1000m.RData")`
--rmcountrycapitals `$(realpath "${HOME}/.nextflow/assets/vmikk/PhyloNext/pipeline_data/CC_Capitals_buf_10000m.RData")`
--rminstitutions `$(realpath "${HOME}/.nextflow/assets/vmikk/PhyloNext/pipeline_data/CC_Institutions_buf_100m.RData")`
--rmurban `$(realpath "${HOME}/.nextflow/assets/vmikk/PhyloNext/pipeline_data/CC_Urban.RData")`
Diversity estimation¶
Diversity estimation is performed using Biodiverse program by Shawn Laffan. Therefore, parameter values should correspond to the Biodiverse values.
| Parameter | Description | Type | Example | Default |
|---|---|---|---|---|
--indices |
Diversity and endemism indices to estimate 1 | string |
"calc_richness,calc_pd,calc_pe" | "calc_richness,calc_simpson_shannon,calc_endemism_whole,calc_pd,calc_pe,calc_phylo_corrected_weighted_endemism,calc_phylo_rpd1,calc_phylo_rpd2,calc_phylo_rpe1,calc_phylo_rpe2" |
--iterations |
Number of randomisation iterations for standardized effect size estimation | integer |
1000 | 1000 |
--biodiversethreads |
Number of Biodiverse threads | integer |
10 | 10 |
--randname |
Randomisation scheme type 2 | string |
"rand_structured" | "rand_structured" |
--randconstrain |
Polygons to perform spatially constrained randomization (GeoPackage format) 2 | file |
ZoogeographicRegions.gpkg |
1:
Comma-separated list of metrics. More than 350 indices are supported.
For details see Biodiverse manual.
The most common indices are listed here: Diversity indices.
2:
In order to estimate standardized effect sizes (SES) of different diversity metrics and
determine whether the obtained estimates are more extreme than what might be predicted given a null model,
Biodiverse requires to perform randomization.
By default, Biodiverse uses the rand_structured randomization algorithm,
which shuffles species occurrences randomly across the entire area while preserving the
species richness of each sample or grid cell. This method works well for most studies and is very effective.
However, in cases where the analysis is conducted on a large scale and involves samples from different biomes or continents,
the total pool of species may span multiple environments. As a result, randomizations may allocate
a polar taxon to the tropics, or a desert taxon to a rainforest. One way to address this issue is
by performing randomizations within a subset of data based on a spatial condition, such as biomes or zoogeographic regions.
PhyloNext allows the user to specify such conditions as a GeoPackage file with multiple polygons.
This feature enables users to keep any species within the biome in which they are found while still randomly relocating them.
Hence, the randomization process can be tailored to suit the specific needs of the study
and ensure that the results accurately reflect the distribution of species in the area of interest.
Randomization algorithms in Biodiverse
See a series of blog posts by Shawn Laffan:
1. Better control of randomisations
2. How the rand_structured algorithm works
3. Spatially partition your randomisations
To illustrate how spatially-constrained randomizations can be executed for Mammals on a global scale,
one can utilize the zoogeographical regions established by Shen et al., 2022, DOI:10.4236/oje.2022.123014.
These regions are conveniently available in the pipeline_data/ZoogeographicRegions.gpkg file,
which is included in PhyloNext.
The file comprises seven biogeographical kingdoms:
- West Palaearctic kingdom
- East Palaearctic kingdom
- Indo-Pacific kingdom
- Afrotropical kingdom
- Australian kingdom
- Nearctic kingdom
- Neotropical kingdom
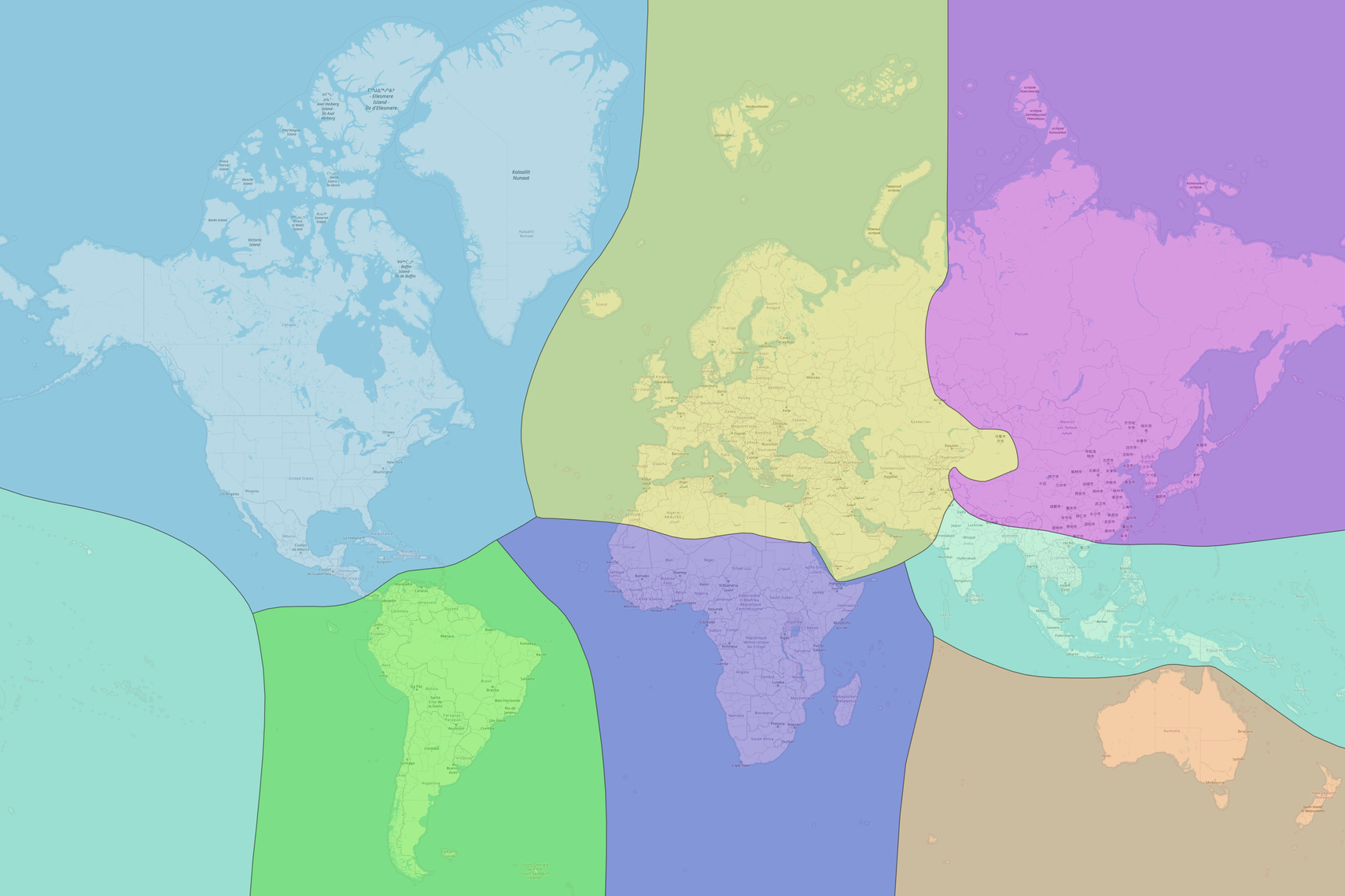
(original data source - https://www.scirp.org/journal/paperinformation.aspx?paperid=116248)
To activate spatially-constrained randomizations using these biogeographic regions, add the following argument to your command:
--randconstrain `$(realpath "${HOME}/.nextflow/assets/vmikk/PhyloNext/pipeline_data/ZoogeographicRegions.gpkg")`
Visualization - interactive¶
Interactive visualization depends on Leaflet library.
| Parameter | Description | Type | Example | Default |
|---|---|---|---|---|
--leaflet_var |
Diversity and endemism indices to estimate 1 | string |
"PD,SES_PD" | "RICHNESS_ALL,PD,SES_PD,PD_P,ENDW_WE,SES_ENDW_WE,PE_WE,SES_PE_WE,PE_CWE,SES_PE_CWE,CANAPE,Redundancy" |
--leaflet_color |
Color scheme for continuous variables 2 | string |
"RdYlBu" | "RdYlBu" |
--leaflet_palette |
Color palette for continuous variables 3 | string |
"quantile" | "quantile" |
--leaflet_bins |
Number of color bins for continuous variables | integer |
5 | 5 |
--leaflet_sescolor |
Color scheme for standardized effect sizes, SES 4 | string |
"hotspots" | "threat" |
--leaflet_redundancy |
Redundancy threshold for hiding the grid cells with low number of records 5 | number |
85 | 0 |
--leaflet_canapesuper |
Include the super-endemism class in CANAPE results 6 |
boolean |
True | False |
1:
For a list of indices available in Biodiverse, see here.
Note: to show the index on a map, please include appropriate name of the Biodiverse subroutine with --indices parameter.
To display Standardized-Effect-Size-based variables, add SES_ prefix to the index name (e.g., SES_PD).
For CANAPE (categorical analysis of neo- and paleoendemism; Mishler et al., 2014), add CANAPE to the variables list.
To display redundancy index (measure of sampling completeness; see Mishler et al., 2020), add Redundancy to the variables list.
2:
The name of a a color scheme from the ColorBrewer designed by Cynthia Brewer.
For more options see here.
3: Defines mapping of data values to colors . See Leaflet docs:
colorNumericis a simple linear mapping from continuous numeric data to an interpolated palettecolorBinalso maps continuous numeric data, but performs binning based on value (see the cut function)colorQuantilesimilarly bins numeric data, but via the quantile function
4:
The default color scheme (threat-type) for SES values follows Mishler et al., 2014,
where high SES values are in blue, and low SES values are in red.
Alternatively, it is possible to use the hotspot-type palette,
where areas with the red values indicate grid cells that contain significantly higher diversity or endemism than expected (hotspots),
while the blue values indicate grid cells that contain significantly lower diversity/endemism than expected (coldspots).
5:
Sampling redundancy is defined as [1 – (richness / number of specimens)] in a grid cell.
By default, parameter --leaflet_redundancy is set to 0, which will display all grid cells.
6: In contrast with the original publication by Mishler et al. (2020), the current version of Biodivere (v.4.1) classifies endemism into 3 categories (super-endemism type is merged with mixed endemism, see https://biodiverse-analysis-software.blogspot.com/2022/10/biodiverse-now-calculates-canape-for-you.html). In PhyloNext, it is possible to use both coloring schemes (with and without super-endemism).
CANAPE
CANAPE (Categorical Analysis of Palaeo and Neo Endemism) is able to distinguish different types of centres of endemism, and can thus give insights into different evolutionary and ecological processes that may be responsible for these patterns.
- The centres of paleo-endemism indicate places where there are over-representation of long branches that are rare across the landscape.
- The centres of neo-endemism indicate an area where there is an over-representation of short branches that are rare on the landscape.
- Mixture of both paleo-endemism and neo-endemism
- Super-endemic sites
Visualization - static¶
| Parameter | Description | Type | Example | Default |
|---|---|---|---|---|
--plotvar |
Variables to plot 1 | string |
"PD" | "RICHNESS_ALL,PD,PD_P" |
--plottype |
Plot type | string |
raw | |
--plotformat |
Plot format (jpg,pdf,png) | string |
||
--plotwidth |
Plot width (default, 18 inches) | number |
18 | |
--plotheight |
Plot height (default, 18 inches) | number |
18 | |
--plotunits |
Plot size units (in,cm) | string |
in | |
--world |
World basemap 2 | file |
enabled |
1: Multiple comma-separated values allowed. For details see --leaflet_var parameter notes.
2:
World base map is based on Natural Earth data
and is located in the pipeline_data/WorldMap_NaturalEarth_Medium.RData file.
Phylogenetic tree-related parameters¶
| Parameter | Description | Type | Default |
|---|---|---|---|
--phylabels |
Type of tip labels on a phylogenetic tree ("OTT" or "Latin") 1 | string |
Latin |
--taxgroup |
Specific taxonomy group in Open Tree of Life (default, "All_life") 2 | string |
All_life |
--maxage |
Manually assign root age for a tree obtained from Open Tree of Life 3 | number |
|
--phyloonly |
Prune Open Tree tips for which there are no phylogenetic inputs 4 | boolean |
false |
1:
If a custom phylogenetic tree was provided by user (with --phytree parameter),
it's important to specify the type of tip labels.
Currently, two labeling schemes are supported:
- Latin binomials in
Genus_speciesformat (e.g., "Homo_sapiens").
NB! whitespaces in the name and sub-species ranks (variety, strain, form, etc.) are not allowed! - Open Tree of Life IDs (e.g., "ott770315")
2:
In the case if tree tips are in OTT-format,
it is possible to limit the query scope at Taxonomic Name Resolution Service of Open Tree of Life.
List available taxonomic contexts in rotl v.3.0.12 package:
- All life (default)
- Animals
- Birds
- Tetrapods
- Mammals
- Amphibians
- Vertebrates
- Arthropods
- Molluscs
- Nematodes
- Platyhelminthes
- Annelids
- Cnidarians
- Arachnids
- Insects
- Land plants
- Hornworts
- Mosses
- Liverworts
- Vascular plants
- Club mosses
- Ferns
- Seed plants
- Flowering plants
- Monocots
- Eudicots
- Rosids
- Asterids
- Asterales
- Asteraceae
- Aster
- Symphyotrichum
- Campanulaceae
- Lobelia
- Fungi
- Basidiomycetes
- Ascomycetes
- Bacteria
- Archaea
- SAR group
- Excavata
- Amoebozoa
- Centrohelida
- Haptophyta
- Apusozoa
- Diatoms
- Ciliates
- Forams
Taxonomic context
Whitespace is not allowed in taxonomic context!
Please replace space with underscore (e.g., "Seed plants" should be speciefied as --taxgroup Seed_plants).
3: In some cases, when there are no age estimates available for the phylogenetic tree nodes, automatic tree retrieval could fail. Therefore, it could be helpful to override the maximum node age to obtain the tree. Note, however, that in this case, units of some of the diversity indices (e.g., PD) become arbitrary.
4:
Open Tree utilizes taxonomic data to augment the structure and completeness of the synthetic tree
when source phylogenies are absent or sparsely sampled (see Hinchliff et al., 2015).
If required, branches lacking phylogenetic support could be removed from the tree using --phyloonly argument.
Open Tree of Life
More information about Open Tree of Life is available at https://opentreeoflife.github.io/.
Generic options¶
| Parameter | Description | Type | Default |
|---|---|---|---|
--deriveddataset |
Export list of GBIF dataset keys for the filtered species occurrences 1 | boolean |
True |
--help |
Display help text (pipeline) | boolean |
|
--helpMsg |
Display help text (pipeline) | boolean |
1: Could be used for citation and preparing a derived dataset with unique DOI.
Nextflow-specific parameters¶
Parameter types
PhyloNext-specific parameters are specified using double dash prefix (e.g., --something value).
Parameters related to the Nextflow workflow manager starts with a single dash prefix (e.g., -work-dir wd)
| Parameter | Description | Type | Example | Default |
|---|---|---|---|---|
-qs |
Queue size (max number of processes that can be executed in parallel) | integer |
8 | number of available CPUs |
-w |
Path to the working directory to store intermediate results | string |
"$(pwd)/wd" | "work" |
-resume |
Execute the pipeline using the cached results. Useful to continue executions that was stopped by an error |
|||
-profile |
Configuration profiles (set of configuration attributes) 1 | string |
test,docker | |
-params-file |
Parameter file in YAML or JSON format 1 | file |
Mammals.yaml |
|
-c |
Configuration file 1 | file |
nextflow.config |
1:
For examples, see the Configuration and profiles and Parameter file sections in the Usage documentation.
Configuration profiles
For more information on Nextflow profiles, see https://www.nextflow.io/docs/latest/config.html#config-profiles.