Troubleshooting¶
Common issues and solutions users might run into while using PhyloNext.
Don't see your error/bug? Post an issue on GitHub
If you've encountered an error or bug not seen here, please post an issue at PhyloNext GitHub Issues. This will help greatly to track down the error and fix it!
Pipeline errors¶
Resume the workflow¶
Pipeline was interrupted
Any accidental failure - e.g., you closed the terminal, rebooted your computer, or the task was killed by a scheduler on HPC.
If the pipeline is interrupted, it can be resumed without having to start from scratch.
Add the -resume flag to your command, and Nextflow will start the workflow from the last successfully executed process,
retrieving all previous results from the cache.
Specifying the full path for input data¶
When inputting data, ensuring the correct file path is essential for successful processing. Incorrect or incomplete paths will lead to errors.
File path
Not a valid path value
To avoid this issue, it is recommended to use the absolute path to your input data. An absolute path specifies the exact location of a file in the file system, starting from the root directory.
For instance, if your file is located in the current working directory,
you can construct its absolute path by prefixing the file name with $(pwd)/.
This command outputs the full path of the current directory, which,
when combined with your file or directory name, provides the complete path required.
Example: $(pwd)/your_file_name.
The number of available CPUs¶
Number of CPUs required
Process requirement exceeds available CPUs -- req: 10; avail: 8
Most likely, the number of CPUs configured for a process is larger than available on your system. To fix it, modify a config file and reduce the number of CPUs for workflow processes to fit your hardware. E.g., change
toSee the Configuration section in the usage documentation.
Pipeline revision version¶
Pipeline revision version
Solution - specify a version number of the pipeline.
To run the latest version, add -r main to your command.
Alternatively, you may specify the exact version (or tag) you wish, e.g. -r v.0.0.2.
Lack of node ages in the phylogenetic tree for target taxon¶
Currently, OpenTree may not contain age estimates for all nodes within the synthetic tree, which can impede the automatic retrieval of a phylogenetic tree for your taxon of interest.
To address this limitation, we outline several strategies below.
Missing age estimate for subtree root
ERROR: no age estimate for subtree root could be found in datastore. Please re-run query with a max root age using argument 'max_age'.
When the database lacks age data for a clade,
you can manually set a maximum age for the clade using the --maxage parameter.
It's advisable to use a credible value, such as one derived from scholarly articles
(e.g., through a Google Scholar search for published age estimates).
Inadequate data for date estimation
ERROR: only 2 or fewer age calibrations found for this tree. We have insufficient data to estimate dates.
In situations with insufficient data for accurate date estimation,
acquiring a custom tree may be necessary.
To derive node age estimates, you can use Chronosynth
and apply Bladj to smooth out the node ages.
For instructions, visit McTavishLab's GitHub repository.
Installation problems¶
Nextflow not found¶
Command not found
command not found: nextflow
If you installed Nextflow in the ~/bin directory, most likely, it is not added to your PATH environment variable.
To fix it, run the following command:
If it works, you may add this command to the ~/.bashrc file to make these changes permanent.
To do it from the command line, run:
Notes on shells other than bash
If you are using a shell other than bash, please modify configuration file specific for your shell.
E.g., for zsh it should be ~/.zshrc.
To know which shell are you using, run echo $SHELL
GitHub API rate limits¶
GitHub API rate limits
Without authentication, GitHub allows only a limited number of connections per hour (60 unauthenticated requests/hr). Most likely, you've hit this problem because users from your organization (e.g., university) send too many requests to GitHub.
To overcome this limitation, you may generate a personal access token at https://github.com/settings/tokens (the minimum required scopes are repo, read:org, and workflow).
To authenticate, you may use the GitHub client gh (https://cli.github.com/).
Run gh auth login --with-token <YOURTOKEN> to login. This way you will have a higher API rate limit.
Alternatively, you may wait for a while until the recovery of API limits.
Or, if you are using PhyloNext locally, you may change your IP address
(for example, connect your laptop to another wi-fi hotspot, e.g., to a smartphone hotspot)
and try to pull the pipeline once again.
You need to download the pipeline from GitHub only once.
Then, Nextflow will cache the pipeline code locally in the ~/.nextflow/assets/ directory
and will re-use it later without the need to connect to GitHub.
As another option, you may just manually clone the git repository into Nextlow assets directory:
mkdir -p ~/.nextflow/assets/vmikk/
cd ~/.nextflow/assets/vmikk/
git clone https://github.com/vmikk/PhyloNext
Rate limit information
To find the current rate limit information (for unauthenticated requests), run
curl -I https://api.github.com/users/octocat
Token security
It is important to keep your token secure and not share it with others.
Also make sure to revoke the token if it is compromised or no longer needed.
Docker permissions¶
Docker - permission denied
docker: Got permission denied while trying to connect to the Docker daemon socket at unix:///var/run/docker.sock: Post "http://%2Fvar%2Frun%2Fdocker.sock/v1.24/containers/create?name=nxf-Cz6yqNjGI9AhRpsqkU4mSAHC": dial unix /var/run/docker.sock: connect: permission denied.
Most likely, you forgot to add your user to the docker group and you need a super-user privileges to execute the container.
To fix this error, run:
# Create the docker group.
sudo groupadd docker
# Add your user to the docker group.
sudo usermod -aG docker ${USER}
# Log out and log back in so that your group membership is re-evaluated.
# Alternatively, to skip logging out, type the following command: su -s ${USER}
# Verify that you can run docker commands without sudo
docker run hello-world
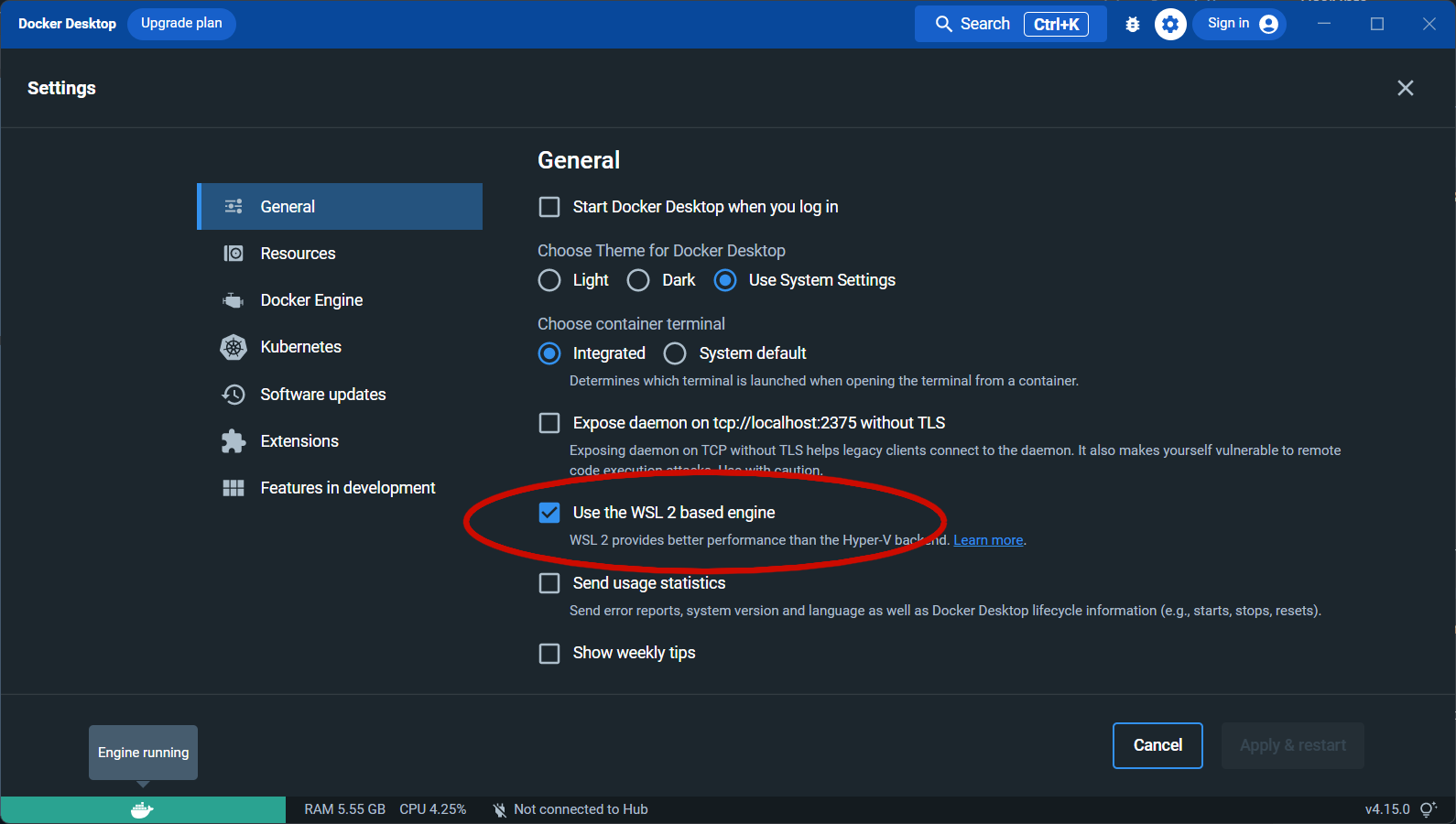
Docker and WSL2¶
Docker and WSL2
The command 'docker' could not be found in this WSL 2 distro. We recommend to activate the WSL integration in Docker Desktop settings.
Try to enable Use the WSL2 based engine in the Docker Desktop settings.
PhyloNext containers¶
If for some reason Nextflow is not able to download containers for PhyloNext, it's possible to pull them manually using: